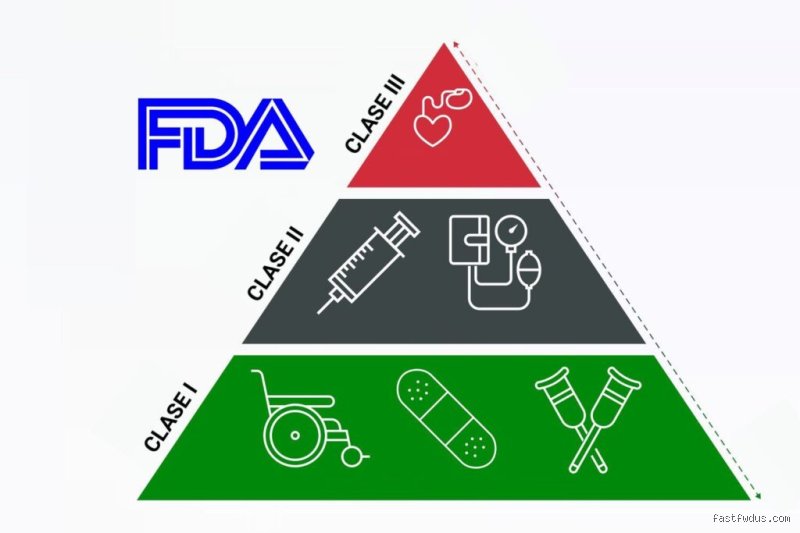
El laberinto de la clasificación: ¿Por qué la FDA nos complica la vida?
Para quienes fabrican tecnología sanitaria, la FDA no es un ente estático, sino un árbitro que juzga la intención de uso y el riesgo intrínseco. No es lo mismo diseñar un termómetro digital que un marcapasos de última generación con inteligencia artificial. La agencia utiliza un sistema basado en el riesgo que divide los dispositivos en tres categorías, pero hoy nos centramos en el salto cuántico que supone pasar del peldaño intermedio al superior. Yo he visto proyectos naufragar porque el equipo de ingeniería subestimó el peso de la evidencia clínica necesaria para saltar esa valla reglamentaria. Pero aquí es donde se complica la narrativa técnica: la clasificación no depende únicamente de la complejidad tecnológica del aparato en cuestión.
La Clase 2: El terreno de los controles especiales
La mayoría de los dispositivos médicos que encuentras en un hospital moderno, aproximadamente el 43% del total regulado, caen en esta categoría de riesgo moderado a alto. Aquí es donde vive el famoso proceso 510(k), esa vía de notificación previa donde el fabricante debe demostrar una equivalencia sustancial con un producto que ya esté legalmente en el mercado. Pero no te equivoques pensando que es un camino de rosas. La Clase 2 impone controles especiales que van desde requisitos de etiquetado específicos hasta estándares de rendimiento obligatorios y una vigilancia post-comercialización que no deja margen al error. ¿Y qué pasa si no hay un predecesor claro? Eso lo cambia todo, obligando a veces a la vía De Novo, pero esa es otra historia.
Clase 3: Donde el riesgo se vuelve una cuestión de supervivencia
Representando apenas un 10% de los dispositivos médicos, la Clase 3 es la joya de la corona y la pesadilla del departamento de cumplimiento. Estamos hablando de tecnologías que sostienen la vida, se implantan de forma permanente o presentan un riesgo potencial irrazonable de enfermedad o lesión. Aquí ya no sirve compararse con el vecino. El fabricante debe presentar una solicitud de PMA (Pre-Market Approval), un expediente que es, esencialmente, una montaña de datos científicos y ensayos clínicos controlados. Porque, al final del día, la FDA necesita una seguridad razonable de que el beneficio supera con creces el peligro de un fallo catastrófico en un dispositivo que el paciente no puede simplemente apagar.
Desarrollo técnico 1: El muro de carga de la Equivalencia Sustancial
Para la Clase 2, el concepto de equivalencia sustancial es el motor que mueve la industria. Si puedes probar que tu monitor de presión arterial o tu bomba de infusión tiene el mismo uso previsto y las mismas características tecnológicas que un dispositivo ya aprobado (el llamado "predicado"), has ganado la mitad de la batalla. Pero, ¡cuidado!, si las tecnologías son distintas, debes demostrar que esas diferencias no plantean nuevas preguntas sobre seguridad y eficacia. Es un baile de matices legales donde un pequeño cambio en el material de un electrodo puede desencadenar una solicitud de información adicional que retrase el lanzamiento 6 meses o más. Y esto ocurre más a menudo de lo que los consultores suelen admitir en sus presentaciones de diapositivas brillantes.
Pruebas de rendimiento y validación en Clase 2
No basta con decir que tu producto funciona; tienes que torturarlo en el laboratorio. Los controles especiales de la Clase 2 a menudo incluyen pruebas de biocompatibilidad, validación de software y pruebas de compatibilidad electromagnética. Imagina que diseñas un escáner de ultrasonidos. Debes cumplir con estándares internacionales como la norma IEC 60601-1, asegurando que el equipo no electrocute al operador ni interfiera con otros sistemas críticos. La documentación es exhaustiva, pero el enfoque sigue siendo comparativo. Muchos creen que la Clase 2 es un trámite ligero, pero la realidad es que el escrutinio sobre la ciberseguridad en dispositivos conectados ha elevado el listón a niveles que hace una década habrían parecido ciencia ficción.
El rigor del etiquetado y el rastreo
Otro pilar técnico fundamental en esta categoría es el sistema de Identificación Única de Dispositivos (UDI). La Clase 2 obliga a un etiquetado que permita el rastreo total desde la fábrica hasta el usuario final. Pero, seamos honestos, esto no es solo por orden administrativo. Si surge un problema de calidad en un lote de jeringas motorizadas, la FDA exige que puedas localizarlas en cuestión de horas. Esta infraestructura logística y de datos suele ser el talón de Aquiles de las startups que saltan de la Clase 1 a la 2 sin haber fortalecido primero su sistema de gestión de calidad bajo la norma 21 CFR Part 820.
Desarrollo técnico 2: La exigencia clínica absoluta de la Clase 3
Cuando cruzamos el umbral hacia la Clase 3, el paradigma cambia del "muéstrame que eres igual a..." al "demuéstrame que esto funciona por sí mismo". La aprobación PMA es, por definición, una revisión científica individualizada. Aquí es donde los datos de ensayos clínicos en humanos se vuelven obligatorios. Estamos lejos de eso que algunos llaman validación rápida. Un estudio clínico para una válvula cardíaca transcatéter puede involucrar a cientos de pacientes en múltiples centros hospitalarios y costar decenas de millones de dólares. La FDA no solo revisa el producto, sino que inspecciona las instalaciones de fabricación antes de emitir cualquier veredicto, asegurándose de que cada unidad producida sea una réplica exacta del prototipo que salvó vidas en el ensayo clínico.
El expediente científico: Más allá del manual de usuario
El volumen de información en un PMA es abrumador. Incluye secciones sobre química, toxicología, inmunología, ingeniería de materiales y, lo más importante, los resultados estadísticos de seguridad y eficacia. Si el valor p de tus resultados clínicos no es lo suficientemente robusto, la agencia simplemente cerrará la puerta. Aquí es donde entra la ironía del sector: a veces, un dispositivo tecnológicamente brillante se queda en el cajón porque el diseño del estudio clínico no fue capaz de aislar las variables necesarias. Estamos hablando de miles de páginas de datos crudos que deben ser digeridos por los revisores del CDRH (Center for Devices and Radiological Health).
Comparación de procesos: El tiempo y el dinero en la balanza
Si analizamos la diferencia entre la Clase 2 y la Clase 3 de la FDA desde una perspectiva puramente operativa, la brecha es un abismo financiero. El tiempo medio de revisión para un 510(k) de Clase 2 ronda los 150 a 180 días, aunque legalmente la FDA apunta a 90. Por el contrario, un PMA de Clase 3 rara vez baja de los 300 a 400 días de revisión activa, sin contar los años previos de investigación clínica. ¿Por qué es relevante esto para nosotros? Porque la estrategia regulatoria dictamina tu capacidad de atraer inversión. Los capitalistas de riesgo suelen huir de la Clase 3 a menos que la innovación sea tan disruptiva que el mercado potencial justifique el riesgo de un "No" rotundo de la agencia tras cinco años de trabajo.
Costes asociados y tasas de la FDA
Hablemos de dinero, porque al final es lo que mantiene las luces encendidas. Las tasas de usuario impuestas por la FDA para el año fiscal vigente reflejan esta disparidad de esfuerzo. Mientras que una solicitud 510(k) para una pequeña empresa puede rondar los $5,000 a $20,000 dólares (dependiendo de las exenciones), un PMA completo puede superar fácilmente los $400,000 dólares solo en tasas gubernamentales. Pero ese es el menor de los problemas; el coste real reside en los salarios de los expertos en asuntos regulatorios y los investigadores principales de los hospitales. Seamos claros: la Clase 3 no es para aficionados ni para presupuestos ajustados.
Mitos desmontados: Errores comunes que hunden proyectos
La falacia de la equivalencia sustancial infinita
Muchos emprendedores creen que la Clase 2 de la FDA es un puerto seguro donde basta con señalar un producto viejo para navegar tranquilo. Error. El problema es que el proceso 510(k) no es una comparativa estética, sino una validación técnica rigurosa de seguridad y eficacia. Y sí, puedes encontrar un dispositivo similar, pero si tu sensor mide la glucosa con un método óptico distinto al del predecesor, la FDA te enviará de vuelta a la casilla de salida. ¿Crees que por ser Clase 2 evitarás los ensayos clínicos? Piénsalo dos veces. Alrededor del 10% de estas solicitudes requieren datos humanos para demostrar que esa supuesta equivalencia no es solo humo y espejos.
El miedo paralizante a la Clase 3
Existe la idea de que entrar en la Clase 3 de la FDA es firmar la sentencia de muerte financiera de una startup. Seamos claros: es un camino tortuoso, caro y desesperadamente lento, pero otorga un foso competitivo casi inexpugnable. No es solo burocracia pesada. Es la diferencia entre un termómetro digital y un marcapasos que mantiene vivo a un abuelo. ¿Por qué íbamos a querer que el proceso fuera ligero? Si tu tecnología es disruptiva y de alto riesgo, la PMA (Premarket Approval) es tu única vía legal. Pero no te engañes, el nivel de escrutinio aquí es tan granular que inspeccionarán hasta el último tornillo de tu planta de fabricación.
¿El software es siempre de bajo riesgo?
La arrogancia del desarrollador de apps es fascinante. Asumen que por ser código y no metal, la diferencia entre Clase 2 y Clase 3 se diluye en una nube de bits. Falso. Un algoritmo que diagnostica un ictus en una tomografía computarizada no es un juguete; es un dispositivo Clase 2 o incluso 3 dependiendo de su autonomía. Si el software decide por el médico, prepárate para un interrogatorio que haría sudar a un espía. Pero, ¿realmente alguien pensó que un código que altera un tratamiento oncológico se aprobaría con un simple formulario web?
El consejo que nadie te da: El De Novo como tierra de nadie
La vía intermedia para los innovadores huérfanos
Si tu dispositivo es de riesgo bajo o moderado (Clase 1 o 2) pero no existe un "predicado" en el mercado, te enfrentas al abismo del proceso De Novo. Es el gran olvidado. Aquí no te comparas con nadie porque eres el primero. Es una oportunidad de oro para establecer las reglas del juego para tus futuros competidores, aunque el coste inicial en documentación sea un trago amargo. Nos gusta ver este camino como el de los valientes que no quieren ser una copia de la copia, sino el nuevo estándar de la industria. No busques un gemelo si tu invento es único; simplemente crea la categoría desde cero y haz que los demás paguen el peaje de seguir tus pasos.
Preguntas Frecuentes sobre la clasificación regulatoria
¿Cuánto cuesta realmente registrar un Clase 2 frente a un Clase 3?
Las cifras marean a cualquiera que no tenga los bolsillos profundos de una multinacional. Para el año fiscal 2024, la tarifa estándar de una solicitud 510(k) para la Clase 2 de la FDA ronda los 21,700 dólares, mientras que una PMA para Clase 3 dispara el termómetro hasta los 483,000 dólares aproximadamente. Pero esto es solo la punta del iceberg administrativo. Debes sumar el coste de los ensayos clínicos, que en dispositivos Clase 3 suelen superar los 5 o 10 millones de dólares fácilmente. La planificación financiera debe ser milimétrica o tu empresa se quedará sin oxígeno antes de recibir la carta de aprobación.
¿Puede un dispositivo Clase 2 ser reclasificado a Clase 3?
La FDA no es una entidad estática y cambia de opinión cuando los datos de seguridad post-mercado revelan sustos inesperados. Ha ocurrido con las mallas quirúrgicas uroginecológicas, que pasaron de una cómoda Clase 2 a la vigilancia extrema de la Clase 3 tras miles de informes de complicaciones graves. (Nadie dijo que el estatus de dispositivo médico fuera eterno). Si tu producto empieza a generar alertas en la base de datos MAUDE, prepárate para una reevaluación que podría obligarte a retirar el stock o realizar estudios clínicos retrospectivos carísimos. Y no, no hay apelación que valga cuando la salud pública está sobre la mesa de los reguladores.
¿Qué papel juega el Control de Diseño en estas categorías?
El Control de Diseño no es una sugerencia amable, es el esqueleto de tu cumplimiento normativo bajo la norma 21 CFR 820.30. Para la Clase 2 y la Clase 3, es obligatorio documentar cada cambio, cada riesgo detectado y cada prueba de validación realizada durante el desarrollo. ¿Sabías que la mayoría de los rechazos iniciales no son por fallos del producto, sino por una documentación mediocre que no sigue la trazabilidad exigida? Si no puedes demostrar con un rastro de papel quién aprobó qué y por qué, tu dispositivo es, a efectos legales, un trozo de plástico inútil. Porque en el mundo de la FDA, lo que no está escrito simplemente no existe.
Síntesis comprometida: Elige tu batalla o abandona el campo
La diferencia entre Clase 2 y Clase 3 no es un mero trámite, sino una decisión existencial que define el ADN de tu negocio. Basta de medias tintas: si buscas rentabilidad rápida y mejoras incrementales, quédate en la Clase 2 y exprime la equivalencia sustancial hasta que no quede nada. Pero si tu ambición es transformar la medicina con tecnologías de soporte vital, abraza el calvario de la Clase 3 con orgullo y presupuesto infinito. Es hipócrita quejarse de la lentitud burocrática cuando lo que estamos evaluando son máquinas que se introducen en el pecho de un ser humano. Al final, la regulación es el único filtro que separa a los visionarios reales de los vendedores de humo con una impresora 3D. Nosotros lo tenemos claro: la seguridad no se negocia, se documenta con sangre, sudor y miles de folios técnicos.